
Nagy, S;
Lau, T;
Alavi, S;
Karimiani, EG;
Vallian, J;
Ng, BG;
Noroozi Asl, S;
Akhondian, J;
Bahreini, A;
Yaghini, O;
et al.
Nagy, S; Lau, T; Alavi, S; Karimiani, EG; Vallian, J; Ng, BG; Noroozi Asl, S; Akhondian, J; Bahreini, A; Yaghini, O; Uapinyoying, P; Bonnemann, C; Freeze, HH; Dissanayake, VHW; Sirisena, ND; Schmidts, M; Houlden, H; Moreno-De-Luca, A; Maroofian, R
(2022)
A recurrent homozygous missense DPM3 variant leads to muscle and brain disease.
Clin Genet, 102 (6).
pp. 530-536.
ISSN 1399-0004
https://doi.org/10.1111/cge.14208
SGUL Authors: Karimiani, Ehsan Ghayoor
|
PDF
Published Version
Available under License Creative Commons Attribution. Download (1MB) | Preview |
|
![[img]](https://openaccess.sgul.ac.uk/id/eprint/115052/6/cge14208-sup-0001-figures1.jpg)
|
Image (JPEG) (Figure S1)
Published Version
Available under License Creative Commons Attribution. Download (186kB) | Preview |
|
![[img]](https://openaccess.sgul.ac.uk/id/eprint/115052/11/cge14208-sup-0002-figures2.jpg)
|
Image (JPEG) (Figure S2)
Published Version
Available under License Creative Commons Attribution. Download (43kB) | Preview |

{kind=link}
{kind=link}
Abstract
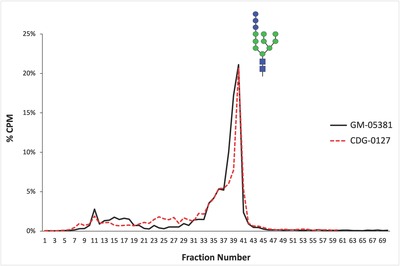
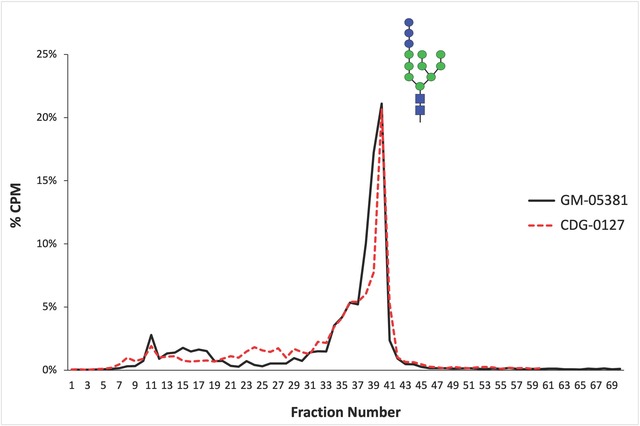
Biallelic pathogenic variants in the genes encoding the dolichol-phosphate mannose synthase subunits (DPM) which produce mannosyl donors for glycosylphosphatidylinositols, N-glycan and protein O- and C-mannosylation, are rare causes of congenital disorders of glycosylation. Pathogenic variants in DPM1 and DPM2 are associated with muscle-eye-brain (MEB) disease, whereas DPM3 variants have mostly been reported in patients with isolated muscle disease-dystroglycanopathy. Thus far, only one affected individual with compound heterozygous DPM3 variants presenting with myopathy, mild intellectual disability, seizures, and nonspecific white matter abnormalities (WMA) around the lateral ventricles has been described. Here we present five affected individuals from four unrelated families with global developmental delay/intellectual disability ranging from mild to severe, microcephaly, seizures, WMA, muscle weakness and variable cardiomyopathy. Exome sequencing of the probands revealed an ultra-rare homozygous pathogenic missense DPM3 variant NM_018973.4:c.221A>G, p.(Tyr74Cys) which segregated with the phenotype in all families. Haplotype analysis indicated that the variant arose independently in three families. Functional analysis did not reveal any alteration in the N-glycosylation pathway caused by the variant; however, this does not exclude its pathogenicity in the function of the DPM complex and related cellular pathways. This report provides supporting evidence that, besides DPM1 and DPM2, defects in DPM3 can also lead to a muscle and brain phenotype.
| Item Type: | Article | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Additional Information: | © 2022 The Authors. Clinical Genetics published by John Wiley & Sons Ltd. This is an open access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0/), which permits use, distribution and reproduction in any medium, provided the original work is properly cited. | ||||||||||||||||||||||||
| Keywords: | DPM3, congenital disorders of glycosylation (CDG), dystroglycanopathy, muscle dystrophy, muscle-eye-brain (MEB) disease, Humans, Intellectual Disability, Homozygote, Muscle, Skeletal, Brain Diseases, Seizures, Mannosyltransferases, Membrane Proteins, Muscle, Skeletal, Humans, Brain Diseases, Seizures, Mannosyltransferases, Membrane Proteins, Homozygote, Intellectual Disability, congenital disorders of glycosylation (CDG), DPM3, dystroglycanopathy, muscle dystrophy, muscle-eye-brain (MEB) disease, 0604 Genetics, 1103 Clinical Sciences, Genetics & Heredity | ||||||||||||||||||||||||
| SGUL Research Institute / Research Centre: | Academic Structure > Molecular and Clinical Sciences Research Institute (MCS) | ||||||||||||||||||||||||
| Journal or Publication Title: | Clin Genet | ||||||||||||||||||||||||
| ISSN: | 1399-0004 | ||||||||||||||||||||||||
| Language: | eng | ||||||||||||||||||||||||
| Publisher License: | Creative Commons: Attribution 4.0 | ||||||||||||||||||||||||
| Projects: |
|
||||||||||||||||||||||||
| PubMed ID: | 35932216 | ||||||||||||||||||||||||
| Web of Science ID: | WOS:000842322700001 | ||||||||||||||||||||||||
| Dates: |
|
||||||||||||||||||||||||
 |
Go to PubMed abstract | ||||||||||||||||||||||||
| URI: | https://openaccess.sgul.ac.uk/id/eprint/115052 | ||||||||||||||||||||||||
| Publisher's version: | https://doi.org/10.1111/cge.14208 |
Statistics
Actions (login required)
 |
Edit Item |